Contenu
- Aperçu
- Signes et symptômes généraux
- Types de maladies du motoneurone
- Diagnostic et traitement
- Faire face
Aperçu
Lorsque vous bougez, des signaux électriques sont envoyés du cerveau à la moelle épinière le long des motoneurones supérieurs. Les cellules nerveuses se synchronisent dans la corne antérieure de la moelle épinière, puis sont envoyées le long des motoneurones inférieurs dans les nerfs périphériques. Les signaux électriques voyageant le long de ces neurones signalent à un muscle de se contracter, entraînant un mouvement.
Les conditions qui affectent cette signalisation normale sont appelées maladies des motoneurones. La corne postérieure de la moelle épinière contient des informations relatives à la sensation, tandis que la corne antérieure porte des informations relatives au mouvement. Les maladies des motoneurones, pour cette raison, affectent principalement le mouvement.
En fonction de certains résultats de l'examen physique, les neurologues peuvent déterminer où se trouve un problème dans le système nerveux et, sur cette base, un diagnostic potentiel.
Signes et symptômes généraux
Les maladies des motoneurones peuvent être séparées en deux catégories principales, selon qu'elles affectent les motoneurones supérieurs ou inférieurs. Certaines maladies des motoneurones n'affectent que les motoneurones supérieurs, tandis que d'autres affectent principalement les motoneurones inférieurs. Certains, comme la SLA, affectent les deux.
Les symptômes de la maladie du neurone moteur supérieur comprennent:
- Spasticité: combinaison de raideur musculaire, d'oppression, de rigidité et d'inflexibilité. Avec une spasticité sévère, vos muscles peuvent se sentir «coincés». Avec une légère spasticité, vous pourrez peut-être bouger vos muscles, mais ils réagissent de manière inattendue ou saccadée.
- Rigidité: Une "raideur" involontaire des muscles.
- Augmentation des réflexes tendineux profonds: par exemple, votre réflexe du genou peut être plus prononcé que d'habitude.
Les symptômes de la maladie du motoneurone inférieur comprennent:
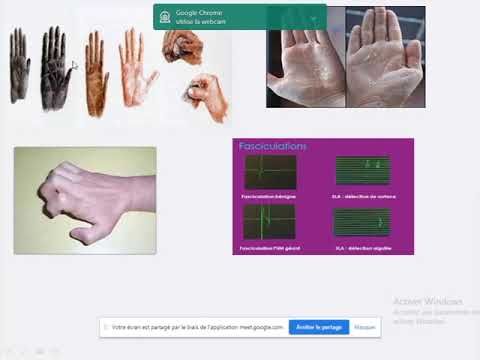
- Atrophie: perte de force et de masse musculaire.
- Fasciculations: contraction spontanée et involontaire des muscles qui peut être visible sous forme de contractions sous la peau.
Types de maladies du motoneurone
Il existe plusieurs maladies des motoneurones qui diffèrent selon qu'elles affectent les motoneurones supérieurs ou inférieurs, les symptômes initiaux, le groupe d'âge qu'elles affectent et le pronostic. Certains d'entre eux incluent:
La sclérose latérale amyotrophique
La sclérose latérale amyotrophique (SLA), également connue sous le nom de maladie de Lou Gehrig, est une maladie des neurones moteurs évolutifs qui touche environ 16 000 Américains. Elle commence par une faiblesse musculaire, généralement d'un seul côté du corps. La condition commence plus souvent dans les mains que dans les pieds. Au début, le signe principal peut être des fasciculations, mais évolue finalement avec des signes et des symptômes des neurones supérieurs et moteurs. Lorsque le diaphragme est affecté, une ventilation mécanique peut être nécessaire.
La maladie n'affecte généralement pas la cognition et la plupart des gens sont alertes (sans démence) même lorsque la maladie est très avancée. L'espérance de vie moyenne avec la SLA est d'environ deux à cinq ans, mais peut varier considérablement, avec 10% des personnes en vie après 10 ans.
Un aperçu de la SLASclérose latérale primaire
La sclérose latérale primaire (PLS) est une maladie des motoneurones supérieurs, perturbant les signaux du cerveau à la moelle épinière. Les cellules du cortex cérébral responsables du mouvement meurent lentement. Le résultat est une faiblesse lentement progressive associée à des signes du motoneurone supérieur, tels que la spasticité, la rigidité et une augmentation des réflexes tendineux profonds. Contrairement à la sclérose latérale amyotrophique, les découvertes des neurones moteurs inférieurs, telles que l'atrophie et les fasciculations, ne sont pas aussi importantes. Il n'est pas certain à quel point le PLS est commun, mais nous pensons qu'il est moins courant que la SLA.
Au début de l'évolution de la maladie, la sclérose latérale primaire peut être confondue avec la SLA. Étant donné que la SLA peut commencer uniquement par des signes du neurone moteur supérieur, il peut s'écouler des années avant qu'un diagnostic de PLS ne devienne apparent. Même à ce moment-là, il peut être difficile de dire laquelle des conditions est à l'origine des symptômes, car certaines personnes atteintes de PLS supposées développeront des découvertes de neurones moteurs inférieurs, ce qui prouve que la maladie est en fait la SLA. Tout cela est une façon assez déroutante de dire qu'il peut être impossible de savoir si une condition est vraiment la SLA ou la PLS pendant plusieurs années après l'apparition des symptômes.
D'autres conditions, telles que la paraparésie spastique héréditaire, devront également être exclues. La PLS a tendance à progresser plus lentement que la SLA, les patients vivant généralement environ une décennie avec leurs symptômes.
Un aperçu de la sclérose latérale primaireAtrophie musculaire progressive
À certains égards, l'atrophie musculaire progressive (PMA) est l'opposé de la sclérose latérale primaire. Dans la PMA, seuls les motoneurones inférieurs sont touchés, tandis que dans le PLS, seuls les motoneurones supérieurs sont lésés. Étant donné que les motoneurones inférieurs sont affectés, la faiblesse progressive est un symptôme courant. Puisque les motoneurones supérieurs ne sont pas affectés, les signes du motoneurone supérieur tels que la rigidité ne se produisent pas. L'atrophie musculaire progressive est moins fréquente que la SLA mais a un meilleur pronostic.
Le diagnostic d'une atrophie musculaire progressive peut être un processus laborieux, car les symptômes sont similaires à ceux d'autres conditions. En particulier, des maladies telles que la SLA, la neuropathie motrice multifocale (une forme de neuropathie périphérique) et l'atrophie musculaire spinale doivent d'abord être exclues avant qu'un diagnostic définitif puisse être posé.
Paralysie bulbaire progressive
La paralysie bulbaire progressive implique une lente dégénérescence du tronc cérébral, qui contient les nerfs (nerfs crâniens) qui contrôlent le visage, la langue et la gorge. En conséquence, une personne atteinte de paralysie bulbaire progressive commencera à avoir des difficultés à parler, à avaler et à mâcher. La faiblesse des membres peut également devenir plus évidente à mesure que la maladie progresse, avec des signes de neurones moteurs supérieurs et inférieurs. Les personnes atteintes de paralysie bulbaire progressive peuvent également avoir des accès de rire ou de pleurs incontrôlables et parfois inappropriés. Il n'est pas rare que les personnes atteintes de paralysie bulbaire progressive développent la SLA. La myasthénie grave est une maladie neuromusculaire auto-immune qui peut également se présenter de manière similaire.
Syndrome post-polio
La polio est un virus qui attaque les motoneurones de la corne antérieure de la moelle épinière, entraînant une paralysie. Heureusement, grâce à des vaccinations agressives, ce virus a été en grande partie éradiqué. Cependant, certains de ceux qui ont eu la maladie peuvent se plaindre d'une faiblesse connue sous le nom de syndrome post-polio. Cela peut être dû au vieillissement ou à une blessure entraînant la mort des quelques motoneurones survivants contrôlant le mouvement d'un membre précédemment affecté. Le trouble ne touche que les personnes âgées qui ont déjà eu la polio. Cela ne met généralement pas la vie en danger.
Maladie de Kennedy
La maladie de Kennedy est due à une mutation génétique liée à l'X qui affecte le récepteur aux androgènes. Le trouble provoque une faiblesse et une douleur lentement progressives des muscles les plus proches du torse. Le visage, la mâchoire et la langue sont également impliqués. Parce qu’elle est liée à l’X, la maladie de Kennedy affecte généralement les hommes. Les femmes porteuses de la mutation génétique sont porteuses, avec 50% de chances de transmettre le gène à leurs enfants. Les femmes atteintes de la mutation peuvent également souffrir de symptômes mineurs, tels que des crampes aux doigts, au lieu d'une faiblesse plus profonde.
Étant donné que la maladie affecte le récepteur aux androgènes (le récepteur auquel les œstrogènes et la testostérone se fixent), les hommes atteints du trouble peuvent également souffrir de symptômes tels que la gynécomastie (augmentation mammaire), l'atrophie testiculaire et la dysfonction érectile. La durée de vie des personnes atteintes de la maladie de Kennedy est généralement normale, bien qu’à mesure que leur faiblesse progresse, elles peuvent avoir besoin d’un fauteuil roulant.
Un aperçu de la maladie de KennedyAtrophie musculaire spinale
L'atrophie musculaire spinale est une maladie héréditaire qui affecte principalement les enfants. Elle est causée par des défauts du gène SMN1 et est héritée selon un schéma autosomique récessif. En raison de ce gène défectueux, une quantité insuffisante de protéines SMN est produite, ce qui conduit à la dégénérescence des motoneurones inférieurs. Cela entraîne une faiblesse et une fonte musculaire.
Il existe trois principaux types de SMA, chacun impliquant des enfants à un âge différent.
- La SMA de type 1, également appelée maladie de Werdnig-Hoffman, devient évidente à l'âge de six mois. L'enfant souffrira d'hypotonie (muscles flasques) et ne bougera pas souvent spontanément. Ils ne pourront pas s'asseoir seuls à l'heure prévue. En raison de difficultés avec les voies respiratoires et de maintenir une force suffisante pour respirer, la plupart de ces enfants meurent à l'âge de deux ans.
- Le SMA de type II commence un peu plus tard, devenant apparent entre 6 et 18 mois. Ces enfants ne pourront ni se tenir debout ni marcher sans assistance et auront également des difficultés respiratoires. Cependant, les enfants atteints de SMA de type II vivent généralement plus longtemps que ceux atteints de Werdnig-Hoffman, vivant parfois à l'âge adulte.
- La SMA de type IIII également appelée maladie de Kugelberg-Welander, apparaît entre l'âge de 2 et 17 ans. Les enfants atteints de ce trouble peuvent avoir des difficultés à courir ou à monter des marches. Ils peuvent également avoir des problèmes de dos, comme une scoliose. Cependant, les enfants atteints de ce trouble peuvent avoir une durée de vie normale.
Diagnostic et traitement
Il n'existe aucun traitement très efficace pour aucune des maladies des motoneurones. La thérapie médicale vise à contrôler au mieux les symptômes de la maladie. Cependant, afin de savoir quels symptômes anticiper, ainsi que d'exclure d'autres maladies plus traitables, il est important d'obtenir le bon diagnostic.
En utilisant leur examen physique et d'autres techniques telles que l'électromyographie, les études de conduction nerveuse et les tests génétiques, le cas échéant, les neurologues peuvent aider à définir le diagnostic correct. Avoir le bon diagnostic permet à votre neurologue de gérer au maximum vos symptômes et d'anticiper et de se préparer à toute complication attendue.
Faire face
Au début, nous avons fait remarquer que les maladies des motoneurones «heureusement» sont rares. Cela peut être bon à moins que vous ou un être cher ne développiez l'une de ces conditions. Ensuite, en plus de souffrir des symptômes de ces maladies, vous constaterez peut-être qu'il y a moins de recherche et moins de soutien que vous ne l'espérez. Bien que ces maladies soient rares, des mesures telles que la loi sur les médicaments orphelins attirent davantage l'attention sur ces conditions moins courantes mais non moins importantes.
Vous pouvez vous sentir seul si vous avez reçu un diagnostic de maladie du motoneurone. Contrairement aux grands groupes de «défenseurs du cancer du sein» là-bas, nous ne voyons pas d'énormes groupes, par exemple, de défenseurs de la paralysie bulbaire progressive. Pourtant, la prise de conscience augmente, et du moins pour la SLA, le soutien.
Les personnes atteintes de maladies des motoneurones ont besoin de soutien, tout comme celles atteintes de maladies plus courantes. Bien que vous n'ayez pas de groupe de soutien dans votre communauté, il sont soutenir les communautés en ligne où les personnes atteintes de troubles neuronaux moteurs spécifiques peuvent «se rencontrer» et communiquer avec d'autres personnes confrontées aux mêmes défis. Bien que nous n'ayons pas de «pilule» ou de chirurgie pour traiter la maladie, il y a beaucoup à faire pour aider les gens à bien vivreavec la maladie, et la recherche actuelle laisse espérer que des progrès seront réalisés dans un avenir pas si lointain.